
A hipertensão arterial pulmonar (HAP) é uma doença rara, com risco de vida elevado se não identificada e tratada adequadamente, caracterizada por uma elevação na resistência vascular pulmonar na ausência de doença ventricular esquerda e aumento da pressão da artéria pulmonar, com conseqüente insuficiência ventricular direita e morte (1). Diversas etiologias têm sido associadas à HAP, incluindo doenças do tecido conjuntivo, cardiopatias congênitas e infecções crônicas, como o HIV. Devido à sua alta prevalência em países em desenvolvimento, uma das formas de HAP mais relevantes em todo o mundo é a associada à esquistossomose.
Epidemiologia
A esquistossomose é a terceira doença parasitária mais prevalente no mundo e, segundo a OMS, há mais de 200 milhões de pacientes contaminados, sendo que em torno de 120 milhões são sintomáticos e 20 milhões apresentam formas graves da doença.
Asa principais regiões do mundo acometidas pela Esquistossomose são África Subsaariana, China, Sudoeste Asiático e algumas áreas da América Latina, sobretudo no Brasil, onde se torna particularmente relevante. Trata-se de uma doença relacionada a pobreza e más condições sanitárias, sendo causada por um verme trematódeo do gênero Schistosoma.
A manifestação mais comum da esquistossomose crônica é a sua forma hepatoesplênica. (2) No entanto, uma das suas manifestações mais graves e limitantes advém do acometimento da circulação pulmonar, com a presença de HAP.
Cerca de 5% dos pacientes com a forma hepatoesplênica da esquistossomose apresentam HAP. (2,3) Assim, dada a alta prevalência mundial da esquistossomose, a HAP-Sch é potencialmente uma das formas mais prevalentes de HAP no mundo, particularmente em países emergentes, podendo representar cerca de 20% dos casos de HAP. (3)
Devido às suas semelhanças histológicas e hemodinâmicas com a HAP idiopática (HAPI), é classificada no grupo 1 (hipertensão pulmonar pré-capilar) da atual classificação da hipertensão pulmonar (4). No entanto, apesar das semelhanças, Sch-PAH tem um curso distinto e mais benigno do que a HAPI, mesmo na ausência de terapia específica (5).
Fisiopatologia:
O pulmão constitui uma etapa obrigatória do ciclo de vida do parasita, que causa lesões pulmonares agudas e crônicas, sendo essas últimas geralmente associadas à forma hepatoesplênica da doença. Do ponto de vista patológico, há evidência de envolvimento arteriolar pulmonar granulomatoso crônico e fibrose, independente da presença dos ovos do parasita na circulação pulmonar. Isso associado ao espessamento na camada média das artérias pulmonares desses pacientes, infiltrado inflamatório na adventícia e a presença de lesões plexiformes (proliferação focal de células endoteliais, com distorção arquitetural secundária) que são semelhantes àquelas encontradas na hipertensão arterial pulmonar idiopática (HAPi). Fig 1 Esse acometimento resulta em remodelamento vascular, aumento da resistência vascular podendo levar à insuficiência do ventrículo direito.
 Fig 1
Fig 1
Quadro Clínico
A apresentação clínica da HAP-Sch é muito semelhante àquela apresentada pelos pacientes portadores de outras formas de HAP. A dispneia progressiva é o principal sintoma. Tontura, síncope e edema periférico estão mais relacionados com a progressão da doença e, hemodinamicamente, com a diminuição do débito cardíaco e consequente insuficiência do ventrículo direito (VD). A dor torácica é um sintoma que também pode estar presente, sendo relacionada com a isquemia crônica do VD ou à compressão coronariana direta pelo tronco da artéria pulmonar. A redução da capacidade de exercício piora progressivamente ao longo do tempo, porém numa progressão mais lenta que a HAPi.
Habitualmente, a radiografia do tórax mostra dilatação importante do tronco da artéria pulmonar e cardiomegalia devido ao aumento do ventrículo direito. O grande aumento das artérias pulmonares, por vezes aneurismáticas, sugere uma característica mais insidiosa dessa patologia. (Fig 2)
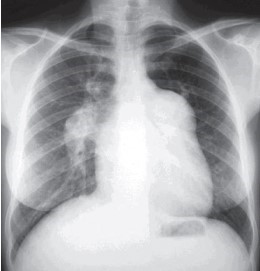 Fig2
Fig2
Diagnóstico
O diagnóstico de HAP-Sch segue o mesmo algoritmo proposto para outras formas de hipertensão arterial pulmonar. Após a suspeita clínica, um ecocardiograma é realizado e, na presença de sinais sugestivos de HP, a confirmação invasiva de HAP através do cateterismo cardíaco de câmaras direitas se faz necessário.
Também se faz necessário que a confirmação de esquistossomose hepatoesplênica seja realizada mediante a presença de alterações ultrassonográficas compatíveis com a doença (presença de fibrose periportal, aumento do lobo caudado e diminuição do lobo direito), acrescido de um dos três fatores epidemiológicos que associem a esquistossomose à condição ultrassonográfica encontrada:
1) Paciente proveniente de área endêmica para a doença;
2) Tratamento prévio para esquistossomose;
3) Presença de ovos do parasita no exame de fezes ou na biópsia retal. (6)
A HAP-Sch possui melhor curso clínico e melhor prognóstico que a HAPI, mesmo na ausência de terapia específica, apresentando melhor sobrevida em três anos do que seria esperado para pacientes com idiopática. Entretanto, isso não faz com que a doença seja isenta de risco e a taxa de mortalidade para esta patologia pode chegar até 15% em 3 anos. (7) Casos de HAP com pronunciada dilatação das artérias pulmonares devem suscitar a hipótese de HAP-Sch, principalmente em regiões altamente prevalentes para essa condição. (8)
Tratamento
O tratamento de HAP-Sch é bastante semelhante ao de outras formas de HAP. Terapia de suporte, com diuréticos e oxigênio deve ser indicada, conforme necessidade, para todos os pacientes com HAP-Sch.
O uso de altas doses de bloqueadores dos canais de cálcio não é aconselhável por causa da presença de hipertensão portal em muitos casos, e ausência de reposta ao teste de vasoreatividade.
Devido à presença frequente de varizes de esôfago nesses pacientes, a anticoagulação deverá ser evitada pelo alto risco de hemorragia digestiva podendo, inclusive, incorrer em óbito.
Todos os pacientes devem receber pelo menos um ciclo do tratamento parasitário, já que não se sabe se a persistência da infecção pode contribuir com a progressão do quadro vascular pulmonar. (3)
Apesar dessa população raramente ser incluída nos grandes ensaios que avaliam terapias direcionadas para HAP (9), a atual diretriz recomenda o tratamento com vasodilatadores pulmonares específicos para a HAP relacionada à esquistossomose, em consonância com outras entidades incluídas no grupo 1 da atual classificação da HP (10). No entanto, a eficácia a longo prazo das terapias direcionadas para HAP ainda precisa ser melhor demonstrada nesse grupo específico.
Referências:
Rubin LJ. Primary pulmonary hypertension. N Engl J Med 1997; 336: 111–117
Gavilanes F, Fernandes CJ, Souza R. Pulmonary Arterial Hypertension in Schistosomiasis. Curr Opin Pulm Med. 2016;22(5):408-14. doi: 10.1097/MCP.0000000000000300.
Fernandes CJC, Piloto B, Castro M, Oleas FG, Alves JL Jr, Prada LFL, et al. Survival of Patients with Schistosomiasis-Associated Pulmonary Arterial Hypertension in the Modern Management Era. Eur Respir J. 2018;51(6):1800307. doi: 10.1183/13993003.00307-2018
Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62: D34–D41.
dos Santos Fernandes CJ, Jardim CV, Hovnanian A, et al.Survival in schistosomiasis-associated pulmonary arterial hypertension. J Am Coll Cardiol 2010; 56: 715–720
Fernandes CJ, Jardim CV, Hovnanian A, Hoette S, Morinaga LK, Souza R. Schistosomiasis and Pulmonary Hypertension. Expert Rev Respir Med. 2011;5(5):675-81. doi: 10.1586/ers.11.58.
Fernandes CJCS, Dias BA, Jardim CVP, Hovnanian A, Hoette S, Morinaga LK, et al. The Role of Target Therapies in Schistosomiasis-Associated Pulmonary Arterial Hypertension. Chest. 2012;141(4):923-8. doi: 10.1378/chest.11-0483.
Hoette S, Figueiredo C, Dias B, Alves JL Jr, Gavilanes F, Prada LFL, et al. Pulmonary Artery Enlargement in Schistosomiasis Associated Pulmonary Arterial Hypertension. BMC Pulm Med. 2015;15:118. doi: 10.1186/s12890-015-0115-y.
Papamatheakis DG, Mocumbi AO, Kim NH, et al.Schistosomiasis-associated pulmonar hypertension. Pulm Circ 2014; 4: 596–611
Galie N, Humbert M, Vachiery JL, et al 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur Respir J 2015; 46: 903–975.





